
Molecular dynamics simulations are crucial for understanding the properties of molecules and materials, with applications in various fields such as drug development and material design. However, simulating the interactions of electrons in molecules has long been a challenging and computationally expensive task.
Traditionally, the interactions of electrons in molecules are computed by solving the Schrödinger equation. This process can be extremely time-consuming, especially for molecules with a large number of atoms. To overcome these limitations, researchers have turned to machine learning methods to predict electronic interactions with reduced computational cost.
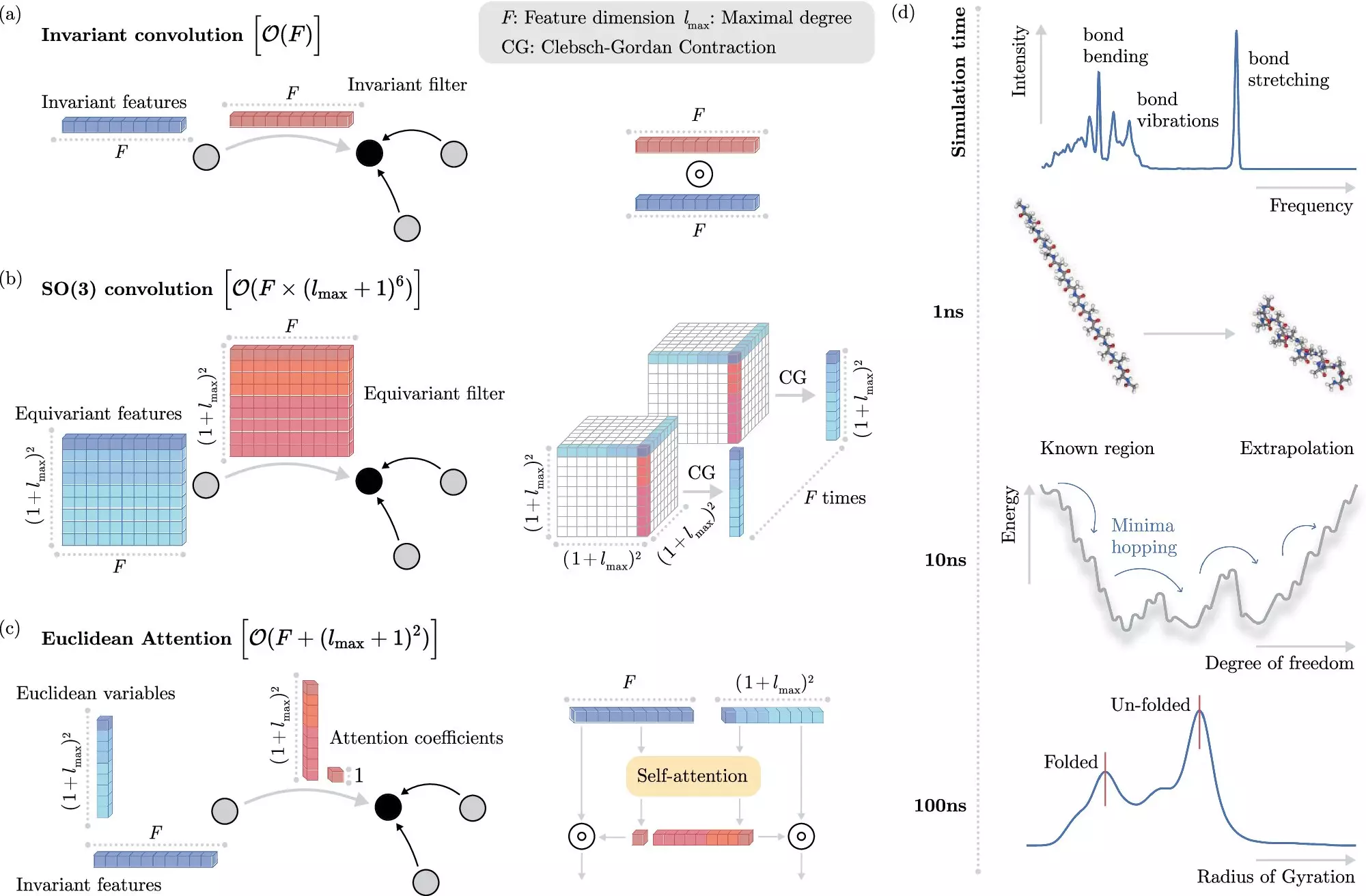
One of the challenges in incorporating machine learning into molecular dynamics simulations is dealing with invariances – properties of molecules that remain the same when molecules are moved in space. While invariances can simplify the learning process, they can also be computationally expensive to implement in machine learning models.
Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) have developed a new machine learning algorithm that addresses the challenge of invariances in molecular dynamics simulations. This algorithm decouples invariances from other information about a chemical system, simplifying the learning process and reducing computational cost.
The new learning algorithm developed by BIFOLD scientists has significantly increased the efficiency of molecular dynamics simulations. Simulations that previously took months or even years to compute can now be completed in a matter of days on a single computer node. This leap in efficiency allows for long-time scale simulations, providing deeper insights into the fundamental processes of nature.
The accurate simulation of molecule-protein interactions could revolutionize drug development by eliminating the need for costly and time-consuming experiments. By using the new machine learning method, researchers were able to identify the most stable version of docosahexaenoic acid, a fatty acid crucial for brain structure, with unprecedented accuracy.
The combination of advanced machine learning techniques with physical principles has the potential to overcome longstanding challenges in computational chemistry. As researchers continue to scale machine learning approaches towards realistic chemical systems, the next generation of algorithms will need to accurately simulate complex, long-range physical interactions.
The integration of machine learning into molecular dynamics simulations has opened up new possibilities for understanding and predicting the behavior of molecules and materials. With continued advancements in machine learning algorithms, the future of computational chemistry looks promising, with the potential to revolutionize drug development, material design, and our understanding of the most complex processes of nature.
Leave a Reply